 |
 |
|
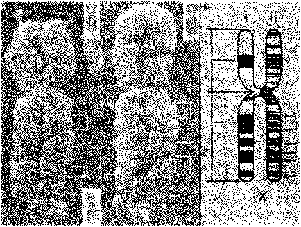
Рис. Микроснимок, на котором запечатлен пробел в хромосоме, который связан с синдромом
хрупкой Х-хромосомы.
Оба эти синдрома (Прадера—Вилли и Энгельмана) связаны с аномалией 15-й хромосомы, но
наследуемыми не признаны. Более того, считается, что они являются спонтанными дефектами
генетического рождения, появляющимися во время зачатия или сразу после него. По причинам, пока не
совсем известным, гены пораженного участка 15-й хромосомы матери являются невыраженными
(функциональными). Этот недостаток гена или генов, тесно примыкающих друг к другу, видимо,
служит причиной указанных синдромов. Происхождение — отцовское или материнское —
отсутствующего генетического материала является возможной причиной явных фенотипических
различий.
Было получено много сведений о генетическом воздействии на интеллект и адаптивные
способности. Поскольку эти воздействия отнюдь не являются единообразными или точно
установленными, остается проблема объяснения механизмов, реализующих эти влияния на интеллект и
вариации выражения фенотипа. Например, даже при синдроме Дауна может встречаться легкая
умственная отсталость, а у некоторых индивидов — наблюдаться нормальный интеллект.
Молекулярные генетические и биологические техники приближают понимание возникновения
подобных вариаций, хотя на сегодняшний день наши знания чрезвычайно ограниченны (State et al.,
1997).
Моногенные формы. Другие синдромы, затрагивающие интеллект и когнитивное
функционирование, могут возникать от метаболических дефектов, имеющих генетическую основу,
которые называются врожденными нарушениями метаболизма. Подобные дефекты вызывают излишек
или недостаток некоторых веществ, необходимых на определенных стадиях развития. Врожденные
нарушения метаболизма объясняют 3-7% случаев тяжелой умственной отсталости (Moser et al., 1990).
Одним из наиболее изученных примеров моногенных форм умственной отсталости является
фенилкетонурия, редкое заболевание, частота которого приблизительно 1 на 10 000 человек (DiLella &
Woo, 1987). В отличие от хромосомных аномалий (являющихся причиной синдрома Дауна), причиной
фенилкетонурии является рецессивный ген, механизмы передачи которого соответствуют законам
Менделя. Дети получают этот ген от обоих родителей — причем, возможно, ни один из них не страдает
фенилкетонурией — следствием чего является недостаток ферментов печени, необходимых для
превращения аминокислоты фенилаланина в другую важнейшую аминокислоту — тирозин. Тирозин в
нормальных условиях превращается в другие вещества, необходимые для физического развития.
Поскольку у больного фенилкетонурией нарушен обмен фенилаланина, присутствующего во многих
продуктах, он накапливается в организме и переходит в фенилпировиноградную кислоту, другой
аномальный метаболит. Этот продукт обмена, в свою очередь, вызывает повреждение мозга,
умственную отсталость, затхлый запах тела, гиперактивность, припадки, а также сухость,
обесцвеченность кожи и волос.
Фенилкетонурия является хорошим примером генетического заболевания, успешно
поддающегося лечению путем изменения окружающей среды. Теперь все новорожденные проверяются
на наличие дефекта и при необходимости немедленно переводятся на строго ограниченную диету.
Однако теперь, благодаря тому что страдающие фенилкетонурией больные получают раннее лечение, у
молодых женщин с этим заболеванием стали появляться дети, что привело к учащению случаев
врожденных дефектов и последующей умственной отсталости у потомков. Строгое ограничение диеты,
введенное до зачатия, в настоящее время является лучшей мерой предупреждения этих проблем